What is Bioinformatics?
Now that you have obtained information about the functional characteristics of alpha amylase, in this exercise you will be comparing the molecular structure of the enzyme in the three species. The tutorial will guide you through finding the gene sequences and comparing them with the BLAST and ClustalW tools.
Bioinformatics is the acquisition, storage, arrangement, identification, analysis, and communication of information related to biology. The term was coined in 1990 with the use of computers in DNA sequence analysis. Think of it as the “theoretical” branch of molecular biology – like the relationship of theoretical physics to the general field of physics.
You will be using the DNA and protein sequence on-line databases that are the core of bioinformatics. There are two general types of sequence databases: Primary databases contain experimental results in an accessible format, but are not sequences that are a population consensus. DDBJ, EMBL, and GenBank are primary databases. Secondary databases are curated to reflect consensus sequences from multiple experiments and usually use the primary databases as their sources.
Abbreviations
DDBJ – DNA Databank of Japan
EMBL – European Molecular Biology Laboratory
NCBI – National Center for Biotechnology Information
BLAST – Basic local alignment search tool
The standard sequence format is called FASTA. All FASTA sequences start with a definition line which consists of:
· a unique identification number (the accession number)
· the version number of the sequence
· the length of the sequence
· molecule type (DNA or mRNA)
· taxonomic division (for instance, INV = invertebrate)
· last release date
· source organism
Every coding sequence also has a unique protein number assigned to it, starting with AA.
Reference sequences (which undergo continuing curation) are the most complete and up-to-date and always start with NT for DNA, NM for mRNA, or NP for protein. Hint – these are the ones you want to use if possible.
Put simply, bioinformatics is the science of storing, retrieving and analysing large amounts of biological information. It is a highly interdisciplinary field involving many different types of specialists, including biologists, molecular life scientists, computer scientists and mathematicians.
The term bioinformatics was coined by Paulien Hogeweg and Ben Hesper to describe "the study of informatic processes in biotic systems" and it found early use when the first biological sequence data began to be shared. Whilst the initial analysis methods are still fundamental to many large-scale experiments in the molecular life sciences, nowadays bioinformatics is considered to be a much broader discipline, encompassing modelling and image analysis in addition to the classical methods used for comparison of linear sequences or three-dimensional structures (Figure 1).
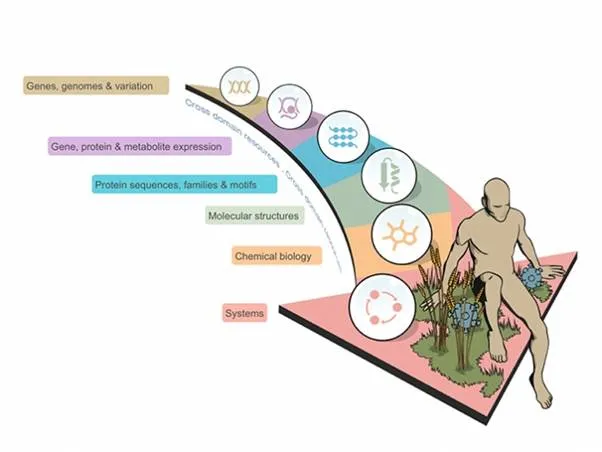
Figure 1 A broad overview of the different types of data that fall within the scope of bioinformatics. Traditionally, bioinformatics was used to describe the science of storing and analysing biomolecular sequence data, but the term is now used much more broadly, encompassing computational structural biology, chemical biology and systems biology (both data integration and the modelling of systems).
Distinction from medical informatics
Bioinformatics is distinct from medical informatics – the interdisciplinary study of the design, development, adoption and application of IT-based innovations in healthcare services delivery, management and planning. Somewhere in between the two disciplines lies biomedical informatics – the interdisciplinary field that studies and pursues the effective uses of biomedical data, information, and knowledge for scientific enquiry, problem solving and decision making, motivated by efforts to improve human health.
Recently initiated projects, such as the 100,000 Genomes Project, are bridging the gaps between these disciplines, but on the whole bioinformatics deals with research data and uses it for research purposes, medical informatics deals with data from individual patients for the purposes of clinical management, (diagnosis, treatment, prevention...) and biomedical informatics attempts to bridge these two extremes.
Sequence Search Introduction
Entrez
Entrez is a data retrieval system developed by the National Center for Biotechnology Information (NCBI) that provides integrated access to a wide range of data domains, including literature, nucleotide and protein sequences, complete genomes, three-dimensional structures, and more. Entrez includes powerful search features that retrieve not only the exact search results but also related records within a data domain that might not be retrieved otherwise and associated records across data domains. These features enable us to gather previously disparate pieces of an information puzzle for a topic of interest. Effective and powerful use of Entrez requires an understanding of the available data domains, the variety of data sources and types within each domain, and Entrez’s advanced search features. This tutorial uses corn (Zea mays) alpha-amlyase to demonstrate the wide variety of information that we can rapidly gather for a single gene. The numbers noted in the search results will of course change over time as the databases grow. The same techniques shown here can be used for any topic of interest.
The search goals are to:
• separate the wheat from the
chaff – identifying a representative, well annotated mRNA or protein sequence
record
• retrieve associated literature
• identify conserved domains within the protein
• identify similar proteins
• find a resolved three-dimensional structure
for the protein or, in its absence, identify structures with homologous
sequence