DNA Damage and Repair
Damage to cellular DNA is involved in mutagenesis and the development of cancer. The DNA in a human cell undergoes several thousand to a million damaging events per day, generated by both external (exogenous) and internal metabolic (endogenous) processes. Changes to the cellular genome can generate errors in the transcription of DNA and ensuing translation into proteins necessary for signaling and cellular function. Genomic mutations can also be carried over into daughter generations of cells if the mutation is not repaired prior to mitosis. Once cells lose their ability to effectively repair damaged DNA, there are three possible responses (see Figure 1).
- The cell may become senescent, i.e., irreversibly dormant. In 2005, multiple laboratories reported that senescence could occur in cancer cells in vivo as well as in vitro, stopping mitosis and preventing the cell from evolving further.
- The cell may become apoptotic. Sufficient DNA damage may trigger an apoptotic signaling cascade, forcing the cell into programmed cell death.
- The cell may become malignant, i.e., develop immortal characteristics and begin uncontrolled division.
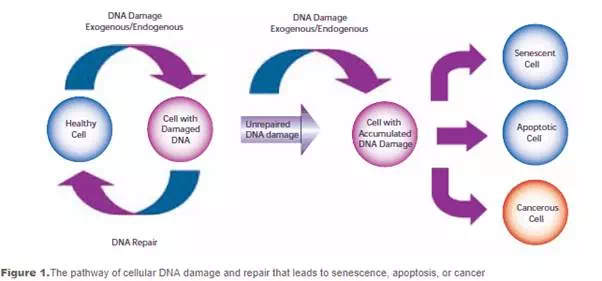
To compensate for the degree and types of DNA damage that occur, cells have developed multiple repair processes including mismatch, base excision, and nucleotide excision repair mechanisms, with little process redundancy. Cells may have evolved to proceed into apoptosis or senescence if overwhelming damage occurs rather than expend energy to effectively repair the damage. The rate at which a cell is able to make repairs is contingent on factors including cell type and cell age.
Sources of DNA Damage
For many years, exogenous sources of damage have been thought to be the primary cause of DNA mutations leading to cancer. However, Jackson and Loeb proposed that endogenous sources of DNA damage also contribute significantly to mutations that lead to malignancy. Both environmental and cellular sources can result in similar types of DNA damage.
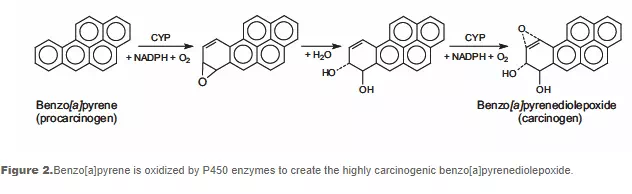
DNA can be attacked by physical and chemical mutagens. Physical mutagens are primarily radiation sources, including UV (200-300 nm wavelength) radiation from the sun. UV radiation produces covalent bonds that crosslink adjacent pyrimidine (cytosine and thymine) bases in the DNA strand. Ionizing radiation (X-rays) initiates DNA mutations by generating free radicals within the cell that create reactive oxygen species (ROS) and result in single-strand and double-strand breaks in the double helix. Chemical mutagens can attach alkyl groups covalently to DNA bases; nitrogen mustard compounds that can methylate or ethylate the DNA base are examples of DNA alkylating agents. Procarcinogens are chemically inert precursors that are metabolically converted into highly reactive carcinogens. These carcinogens can react with DNA by forming DNA adducts, i.e., chemical entities attached to DNA. Benzo[a]pyrene, a polyaromatic heterocycle, is not itself carcinogenic. It undergoes two sequential oxidation reactions mediated by cytochrome P450 enzymes, which results in benzo[a]pyrenediol epoxide (BPDE), the carcinogenic metabolite that is able to form a covalent DNA adduct (see Figure 2).
DNA damage can also result from endogenous metabolic and biochemical reactions, some of which are not well understood.6Hydrolysis reactions can partially or completely cleave the nucleotide base from the DNA strand. The chemical bond connecting a purine base (adenine or guanine) to the deoxyribosyl phosphate chain can spontaneously break in the process known as depurination. An estimated 10,000 depurination events occur per day in a mammalian cell.7 Depyrimidination (loss of pyrimidine base from thymine or cytosine) also occurs, but at a rate 20 to 100-fold lower than depurination.
Deamination occurs within the cell with the loss of amine groups from adenine, guanine, and cytosine rings, resulting in hypoxanthine, xanthine, and uracil, respectively. DNA repair enzymes are able to recognize and correct these unnatural bases. However, an uncorrected uracil base may be misread as a thymine during subsequent DNA replication and generate a C→T point mutation.
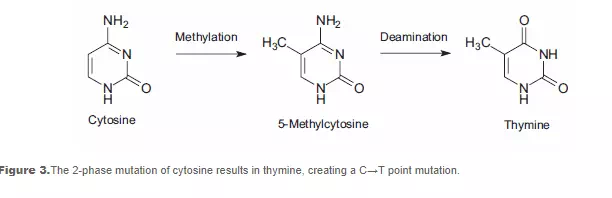
DNA methylation, a specific form of alkylation, occurs within the cell due to a reaction with S-adenosyl methionine (SAM). SAM is an intracellular metabolic intermediate that contains a highly reactive methyl group. In mammalian cells, methylation occurs at the 5-position of the cytosine ring of a cytidine base (C) that is 5’ to a guanosine base (G), i.e., sequence CpG. A significant source of mutation error is the spontaneous deamination of the 5-methylcytosine product of methyl-ation. Loss of the amine group results in a thymine base, which is not detected by DNA repair enzymes as an unnatural base. The resulting substitution is retained in DNA replication, creating a C→T point mutation (see Figure 3).
Normal metabolic processes generate reactive oxygen species (ROS), which modify bases by oxidation. Both purine and pyrimidine bases are subject to oxidation. The most common mutation is guanine oxidized to 8-oxo-7,8-dihydroguanine, resulting in the nucleotide 8-oxo-deoxy guano sine (8-oxo-dG). The 8-oxo-dG is capable of base pairing with deoxyadenosine, instead of pairing with deoxycytotidine as expected. If this error is not detected and corrected by mismatch repair enzymes, the DNA subsequently replicated will contain a C→A point mutation. ROS may also cause depurination, depyrimidination, and single-strand or double strand breaks in the DNA.
Other genomic mutations may be introduced during DNA replication in the S phase of the cell cycle. Polymerases that duplicate template DNA have a small but significant error rate, and may incorporate an incorrect nucleotide based on Watson-Crick pairing versus the template DNA. Chemically altered nucleotide precursors may be incorporated into the generated DNA by the polymerase, instead of normal bases. In addition, polymerases are prone to “stuttering” when copying sections of DNA that contain a large number of repeating nucleotides or repeating sequences (microsatellite regions). This enzymatic “stuttering” is due to a strand slippage, when the template and replicated strands of DNA slip out of proper alignment. As a result, the polymerase fails to insert the correct number of nucleotides indicated by the template DNA, resulting in too few or too many nucleotides in the daughter strand.
Single strand and double strand cleavage of the DNA may occur. Single strand breaks may result from damage to the deoxyribose moiety of the DNA deoxyribosylphosphate chain. Breaks also result as an intermediate step of the base excision repair pathway after the removal of deoxyribose phosphate by AP-endonuclease
1. When a single strand break occurs, both the nucleotide base and the deoxyribose backbone are lost from the DNA structure. Double strand cleavage most often occurs when the cell is passing through S-phase, as the DNA may be more susceptible to breakage while it is unraveling for use as a template for replication.
Mechanisms of DNA Repair
While the cell is able to evolve into either an apoptotic or senescent state, these actions are performed as a last resort. For each type of DNA damage, the cell has evolved a specific method of repairing the damage or eliminating the damaging compound.
O6-Methylguanine DNA methyltransferase (MGMT; DNA alkyltransferase) cleaves both methyl and ethyl adducts from guanine bases on the DNA structure. The reaction is not a catalytic (enzymatic) reaction but is stoichiometric (chemical), consuming one molecule of MGMT for each adduct removed. Cells that have been engineered to overexpress MGMT are more resistant to cancer, likely because they are able to negate a larger amount of alkylating damage. A recent study by Niture, et al., reports an increase in MGMT expression by use of cysteine/glutathione enhancing drugs and natural antioxidants.
DNA polymerases such as polymerase-δ contain proofreading activities and are primarily involved in replication error repair. When an error is detected, these polymerases halt the process of DNA replication, work backward to remove nucleotides from the daughter DNA chain until it is apparent that the improper nucleotide is gone, and then reinitiate the forward replication process. Mice with a point mutation in both copies of the Pold1 gene demonstrated a loss of proofreading activity by DNA polymerase-δ and developed epithelial cancers at a significantly higher rate than did mice with wild-type genes or with a single copy mutation.
A group of proteins known as mismatch excision repair (MMR) enzymes is capable of correcting errors of replication not detected by the proofreading activities of DNA polymerase. MMR enzymes excise an incorrect nucleotide from the daughter DNA and repair the strand using W-C pairing and the parent DNA strand as the correct template. This is especially crucial for errors generated during the replication of microsatellite regions, as the proofreading activity of DNA polymerase does not detect these errors. To a lesser degree, MMR enzymes also correct a variety of base pair anomalies resulting from DNA oxidation or alkylation. These mutations include modified base pairs containing O6-methylguanine and 8-oxoguanine, and carcinogen and cisplatin adducts. Mutations in the human mismatch excision repair genes MSH2 and MLH1 are associated with hereditary non-polyposis colorectal cancer (HNPCC) syndrome.
Base Excision Repair and Nucleotide Excision Repair
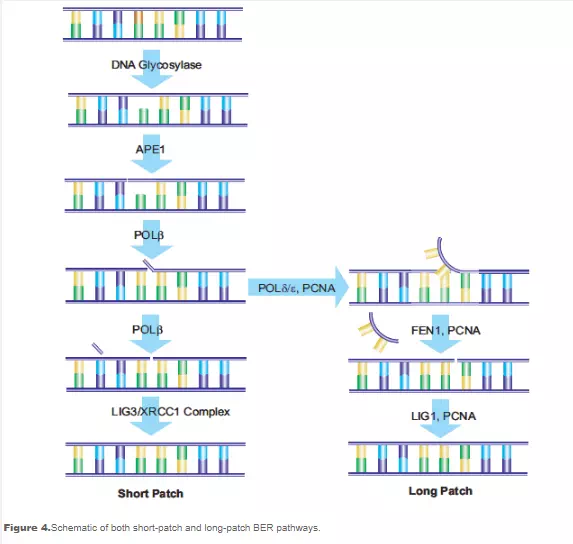
Base excision repair (BER) involves multiple enzymes to excise and replace a single damaged nucleotide base. The base modifications primarily repaired by BER enzymes are those damaged by endogenous oxidation and hydrolysis. A DNA glycosylase cleaves the bond between the nucleotide base and ribose, leaving the ribose phosphate chain of the DNA intact but resulting in an apurinic or apyrimidinic (AP) site. 8-Oxoguanine DNA glycosylase I (Ogg1) removes 7,8-dihydro-8-oxoguanine (8-oxoG), one of the base mutations generated by reactive oxygen species. Polymorphism in the human OGG1 gene is associated with the risk of various cancers such as lung and prostate cancer. Uracil DNA glycosylase, another BER enzyme, excises the uracil that is the product of cytosine deamination, thereby preventing the subsequent C→T point mutation. N-Methylpurine DNA glycosylase (MPG) is able to remove a variety of modified purine bases.
The AP sites in the DNA that result from the action of BER enzymes, as well as those that result from depyrimidination and depurin ation actions, are repaired by the action of AP-endonuclease 1 (APE1). APE1 cleaves the phosphodiester chain 5’ to the AP site. The DNA strand then contains a 3’-hydroxyl group and a 5’-abasic deoxyribose phosphate. DNA polymerase β (Polβ) inserts the correct nucleotide based on the corresponding W-C pairing and removes the deoxyribose phosphate through its associated AP-lyase activity. The presence of X-ray repair cross-complementing group 1 (XRCC1) is necessary to form a heterodimer with DNA ligase III (LIG3). XRCC1 acts as a scaffold protein to present a non-reactive binding site for Polβ, and bring the Polβ and LIG3 enzymes together at the site of repair.17 Poly(ADP-ribose) polymerase (PARP-1) interacts with XRCC1 and Polβ and is a necessary component of the BER pathway.18,19 The final step in the repair is performed by LIG3, which connects the deoxyribose of the replacement nucleotide to the deoxyribosylphosphate backbone. This pathway has been named “short-patch BER”.20
An alternative pathway called “long-patch BER” replaces a strand of nucleotides with a minimum length of 2 nucleotides. Repair lengths of 10 to 12 nucleotides have been reported. Longpatch BER requires the presence of proliferation cell nuclear antigen (PCNA), which acts as a scaffold protein for the restructuring enzymes. Other DNA polymerases, possibly Polδ and Polε, are used to generate an oligonucleotide flap. The existing nucleotide sequence is removed by flap endonuclease-1 (FEN1). The oligonucleotide is then ligated to the DNA by DNA ligase I (LIG1), sealing the break and completing the repair. The process used to determine the selection of short-patch versus long patch BER pathways is still under investigation (see Figure 4).
While BER may replace multiple nucleotides via the long-patch pathway, the initiating event for both short-patch and long-patch BER is damage to a single nucleotide, resulting in minimal impact on the structure of the DNA double helix. Nucleotide excision repair (NER) repairs damage to a nucleotide strand containing at least 2 bases and creating a structural distortion of the DNA. NER acts to repair single strand breaks in addition to serial damage from exogenous sources such as bulky DNA adducts and UV radiation. The same pathway may be used to repair damage from oxidative stress. Over 20 proteins are involved in the NER pathway in mammalian cells, including XPA, XPC-hHR23B, replication protein A (RPA), transcription factor TFIIH, XPB and XPD DNA helicases, ERCC1-XPF and XPG, Polδ, Polε, PCNA, and replication factor C. Overexpression of the excision repair cross-complementing (ERCC1) gene has been associated with cisplatin resistance by non-small-cell lung cancer cells and corresponds to enhanced DNA repair capacity. Global genomic NER (GGR) repairs damage throughout the genome, while a specific NER pathway called Transcription Coupled Repair (TCR) repairs genes during active RNA polymerase transcription.
Repair of Double-Strand Breaks
Double-strand breaks in DNA can result in loss and rearrangement of genomic sequences. These breaks are repaired by either nonhomologous end-joining (NHEJ) or by homologous recombination (HR), also called recombinational repair or template–assisted repair.
The HR pathway is activated when the cell is in late S/G2 phase and the template has recently been duplicated. This mechanism requires the presence of an identical or nearly identical sequence linked to the damaged DNA region via the centromere for use as a repair template. Double-stranded breaks repaired by this mechanism are usually caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion, resulting in the collapse of the replication fork.
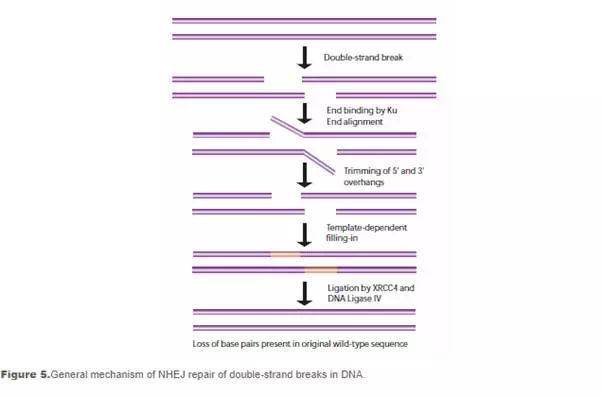
Non-homologous end-joining (NHEJ) is used at other points of the cell cycle when sister chromatids are not available for use as HR templates. When these breaks occur, the cell has not yet replicated the region of DNA that contains the break, so unlike the HR pathway, there is no corresponding template strand available. In NHEJ, the Ku heterodimeric protein positions the two ends of the broken DNA strands for repair without an available template, losing sequence information in the process. Multiple enzymes are involved in the rejoining process, including DNA ligase IV, XRCC4, and DNA-dependent protein kinase (DNA-PK). NHEJ is inherently mutagenic as it relies on chance pairings, called microhomologies, between the single-stranded tails of the two DNA fragments to be joined (see Figure 5). In higher eukaryotes, DNA-PK is required for NHEJ repair, both via the primary mechanism and via an alternative back-up mechanism (D-NHEJ).34
Future Applications
While DNA damage is a key factor in the development and evolution of cancer cells, continued damage is used as part of clinical treatments for cancer, forcing malignant cells into apoptosis or senescence. Many chemotheraputic drugs such as bleomycin, mitomycin, and cisplatin, are effective because they cause further DNA damage in cancer cells that replicate at a faster rate than surrounding tissue. Cellular DNA repair mechanisms are a doubleedged sword; by reducing mutations that may lead to cancer, these processes strive for genomic integrity, but the same mechanisms in malignant cells allow those cells to survive additional DNA damage and continue uncontrolled growth. In order to block this survival mechanism within cancer cells, clinical trials are now being performed using inhibitors to specific DNA repair enzymes, including MGMT, PARP, and DNA-PK.